
Identification of Natural GPVI Receptor Antagonists Using Molecular Docking and Molecular Dynamics
Cardiovascular diseases are frequently associated with abnormal platelet aggregation leading to thrombus formation. Glycoprotein VI (GPVI) is a collagen receptor expressed on platelet membranes that plays a central role in collagen-induced platelet activation and thrombus formation. Targeting GPVI represents a promising therapeutic strategy for antithrombotic drug development because inhibiting this receptor may reduce thrombosis risk while minimizing bleeding complications associated with traditional antiplatelet drugs.
This project explored the potential of naturally occurring flavonoids from Citrus limon as small-molecule antagonists of GPVI using a computational drug discovery pipeline combining structural bioinformatics, molecular docking, interaction analysis, and molecular dynamics simulations.
Structural Analysis of GPVI
The three-dimensional structure of the human platelet GPVI receptor (PDB ID: 2GI7) was retrieved from the Protein Data Bank and analyzed to identify potential ligand binding pockets.
Figure – GPVI Receptor Structure
Binding site prediction identified several pockets within the receptor structure. Key residues including Lys59, Arg60, and Arg166 were identified as critical for collagen binding and were therefore selected as target residues for docking analysis.
Understanding the spatial organization of these binding pockets enabled the identification of regions that could accommodate potential inhibitory ligands.
Ligand Library Construction
A library of 23 flavonoid compounds with reported antiplatelet, antioxidant, and anti-atherogenic properties was compiled using phytochemical databases.
Examples of screened compounds include:
- Eriodictyol
- Luteolin
- Hesperetin
- Rutin
- Vitexin
- Quercetin
- Diosmetin
- Didymin
Three-dimensional ligand structures were obtained from PubChem, converted into docking-compatible formats using Open Babel, and prepared for docking simulations.
Molecular Docking
Molecular docking simulations were performed using AutoDockTools, which evaluates ligand binding by predicting optimal conformations and calculating binding energy scores.
The docking procedure involved:
- Preparation of the receptor by removing water molecules and adding hydrogen atoms
- Generation of grid maps surrounding the active site
- Application of a genetic algorithm to search ligand conformational space
- Evaluation of binding energies and interaction profiles
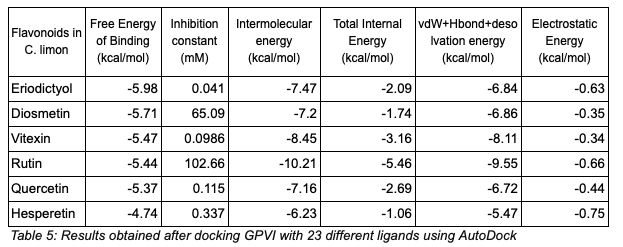
The docking results were ranked based on free binding energy, hydrogen bonding interactions, and hydrophobic contacts.
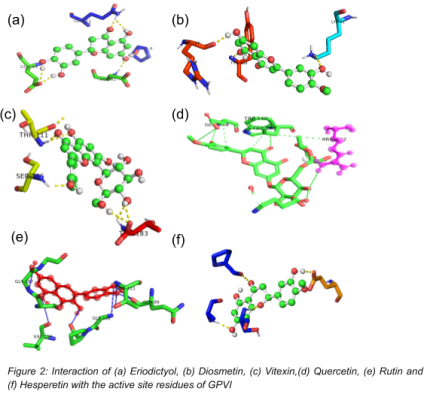
Figure – Protein–Ligand Docking Interactions
Several ligands showed strong interactions with the GPVI binding site, particularly through hydrogen bonding and hydrophobic contacts with key residues.
Protein–Ligand Interaction Analysis
The docked complexes were analyzed using the Protein-Ligand Interaction Profiler (PLIP) to identify key intermolecular interactions.
The analysis revealed:
- Multiple hydrogen bond interactions with residues such as Glu84 and Arg166
- Hydrophobic contacts with residues including Leu17, Val52, and Trp140
- Additional stabilizing interactions including π-stacking and electrostatic interactions
These interactions contribute to the stability and specificity of ligand binding within the receptor pocket.
Drug-Likeness and Biological Activity Prediction
Top-ranked compounds were further evaluated using PASS and MolSoft tools to assess predicted biological activity and drug-likeness.
Evaluated properties included:
- Molecular weight
- Hydrogen bond donors and acceptors
- LogP and solubility
- Polar surface area
- Blood–brain barrier permeability
- Drug-likeness scores
Among all candidates, Eriodictyol demonstrated the most favorable drug-likeness score and binding profile.
Molecular Dynamics Simulation
To further evaluate the stability of the protein-ligand complex, Molecular Dynamics (MD) simulations were conducted using GROMACS with the CHARMM36 force field.
Simulation workflow included:
- Energy minimization
- NVT and NPT equilibration
- Production simulation runs
- Analysis of thermodynamic parameters
Figure – Energy Minimization and System Stability
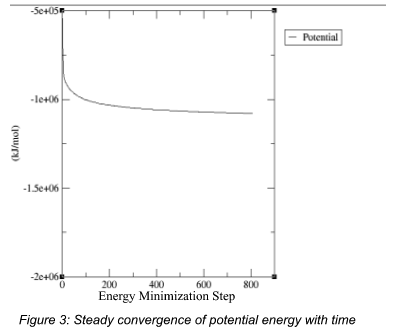
Figure – Temperature Stabilization During Simulation
Figure – Density Equilibration
The MD simulations demonstrated:
- Stable temperature convergence (~297 K)
- Stable density (~1011 kg/m³)
- Low RMSD fluctuations indicating structural stability
The GPVI–Eriodictyol complex remained stable throughout the simulation, supporting the docking results.
Key Findings
- Several flavonoids exhibited strong binding affinity toward the GPVI receptor.
- Eriodictyol demonstrated the best binding profile with favorable docking energy and drug-likeness properties.
- Interaction analysis confirmed stable hydrogen bonding and hydrophobic contacts with GPVI active site residues.
- Molecular dynamics simulations validated the structural stability of the GPVI–Eriodictyol complex.
These results suggest that Eriodictyol may act as a promising natural GPVI antagonist, supporting the potential of plant-derived compounds for antithrombotic drug discovery.