Unraveling the binding potential of natural lead molecules from Rosmarinus officinalis and Aegle marmelos towards multiple targets of SARS CoV-2 by computer aided virtual screening and cheminformatics approaches
- 7 minsDayananda Sagar College of Engineering, India. Summer 2020
Background
In light of the current absence of approved drugs against COVID-19, computational biology tools have been harnessed to identify promising leads against SARS CoV-2. The plant-based bioactive compounds found in Rosmarinus officinalis and Aegle marmelos have been noted for their antioxidant, antimicrobial, anticancer, and anti-inflammatory properties, prompting researchers to explore their potential as therapeutic agents. The goal of this study was to employ computer-aided virtual screening to identify natural compounds that could target multiple SARS-CoV-2 proteins. To achieve this, sixty-five natural compounds from Rosmarinus officinalis and Aegle marmelos were screened for drug-like properties and pharmacokinetics, with a focus on the membrane protein, Spike RBD, 3CLpro, PLpro, and RdRp as primary targets due to their virulent functions. Since the three-dimensional structure of the membrane protein was unavailable in its native form, a computational model was created. Molecular docking was used to study the binding potential of the selected natural molecules to these targets, and the interaction was compared to the FDA-approved drug Favipiravir. The stability of the best docked complexes was further validated through molecular dynamic (MD) simulation. Four natural compounds, namely Genkwanin, Psoralen, gamma-Fagarine, and Dictamine, showed significant binding potential to the prioritized targets in comparison with the control, demonstrating their promise as lead molecules against SARS-CoV-2. The MD simulation confirmed the stability of the complexes throughout the interaction. This study highlights the potential of these natural molecules as therapeutic agents against multiple targets of SARS-CoV-2, and provides a roadmap for further experimental validation.
Methods
- Molecular target identification from SARS CoV-2
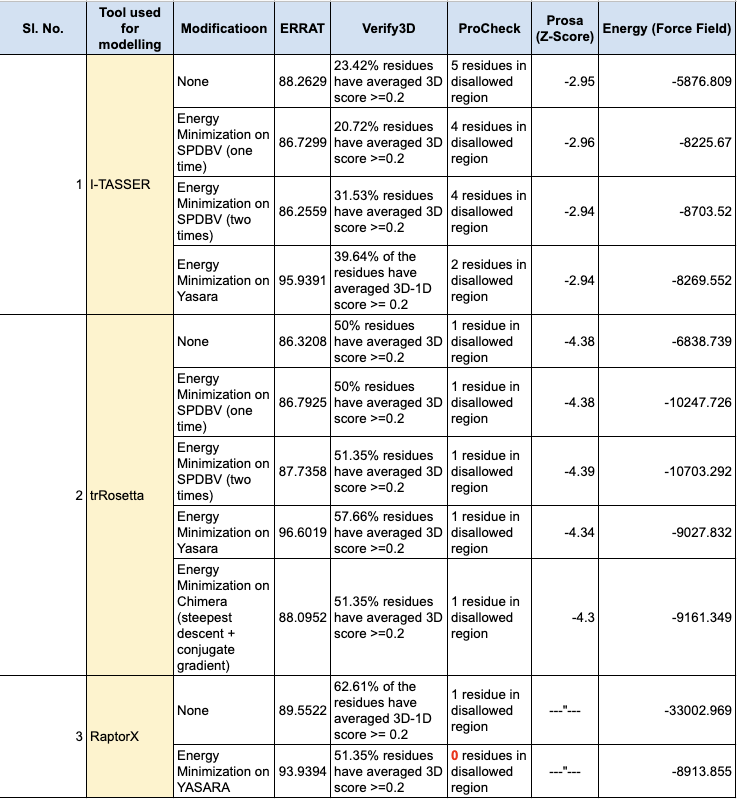
The literature survey revealed that key proteins in SARS CoV-2 responsible for causing the disease included the spike receptor binding domain, 3C-like protease (3CLPro), papain-like protease (PLpro), RNA dependent RNA polymerase (RdRp), and membrane protein. Structures for four of these proteins were retrieved from RCSB-PDB, including the spike receptor-binding domain (PDB ID: 6M0J), 3CLpro (PDB ID: 6LU7), PLpro (PDB ID: 6W9C), and RdRp (PDB ID: 6M71). However, the native structure of the membrane protein was not available. To address this, the sequence for the membrane protein was obtained from the UniProt database (Uniprot ID: A0A6C0NA72), and a hypothetical model was generated using the trRosetta online tool, which utilizes deep neural networks. The secondary structure of the protein was predicted using SOPMA, and the hypothetical model was computationally validated using ERRAT, Verify3D, ProCheck, and Prosa. Overall, the model was assessed for its quality, stereochemical quality, and three-dimensional structure errors using these tools.
- Identification of natural lead molecules through virtual screening
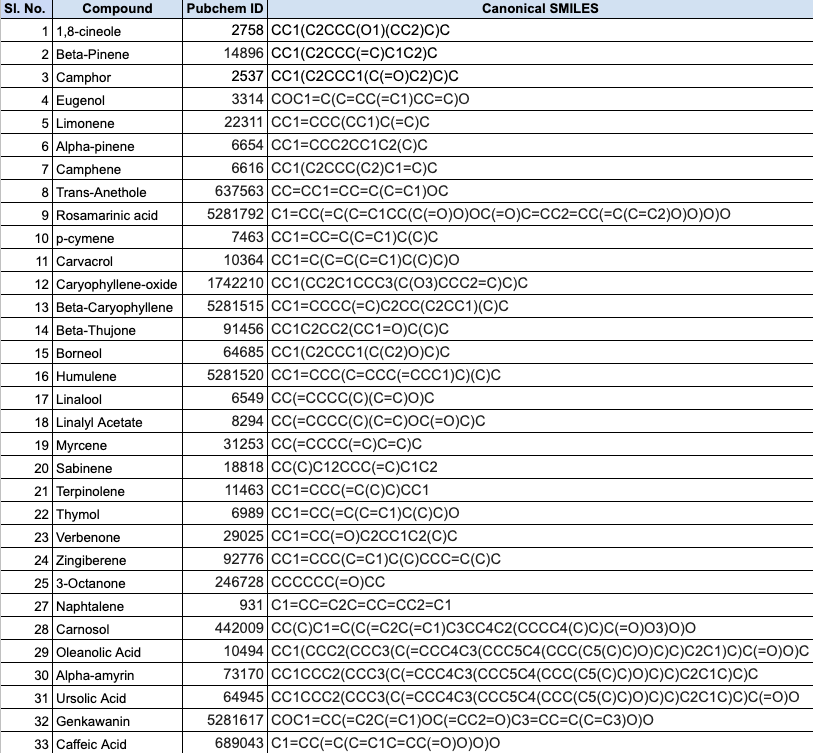
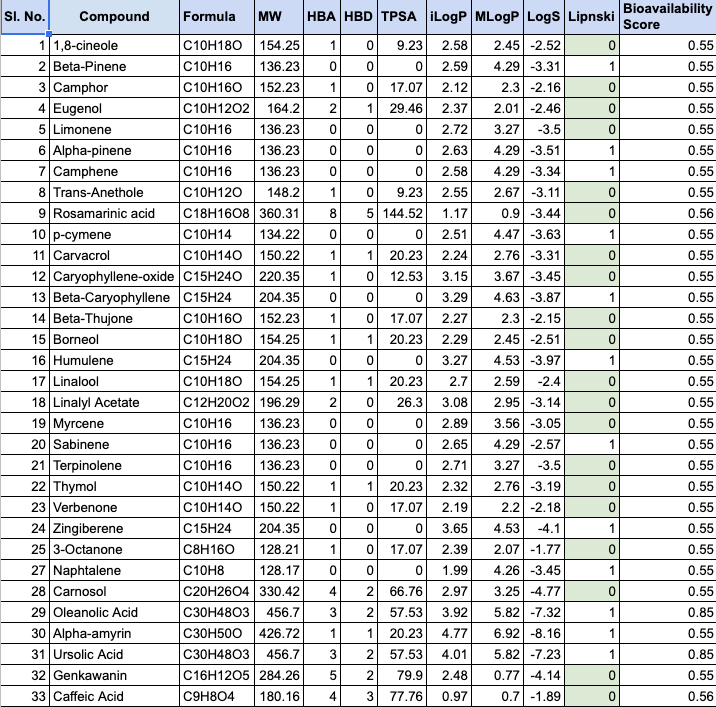
Further, we proceeded to perform the identification and screening of natural compounds from two plant sources, R. officinalis and A. marmelos, for their potential antiviral properties. Around 65 natural compounds were identified and their three-dimensional structures were obtained from the PubChem database and converted to .pdb format using Open Babel. The drug likeness, pharmacokinetic, and toxicity properties of the identified compounds were studied using PreADMET and SwissADME, which applied various molecular filters and parameters to assess their drug likeness, ADME properties, and toxicity. Only the compounds that met the criteria were selected for further studies. The table below lists the top 33 potential ligands.
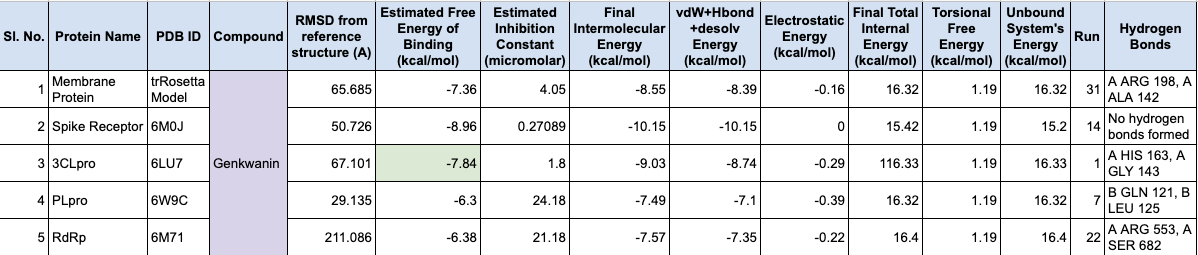
- Molecular docking studies
The next step was to predict the binding potential of natural compounds against SARS-CoV-2 molecular targets. This includes literature survey to identify potential molecular targets, natural compounds and their sources, obtaining their 3D structures, studying their drug-likeness, pharmacokinetic and toxicity properties, and predicting the binding sites of molecular targets using CastP tool. You have also mentioned the use of Autodock tools and Autodock Vina for multiple ligand docking, and the use of MGL tools for molecular docking simulation. The results were compared with the binding potential of a control, Favipiravir. This methodology provides a comprehensive approach to identify potential natural compounds for drug development against SARS-CoV-2.
- Molecular dynamic simulation of the best docked complexes
To gain insight into the stability and dynamics of the protein-ligand complexes identified through docking, molecular dynamics (MD) simulations were performed using the Desmond module of Schrödinger’s software suite. The simulations were run for 100 nanoseconds, during which the system was maintained at a constant temperature, pressure, and number of particles (NPT) to ensure accuracy and reliability. The root mean square deviation (RMSD) and root mean square fluctuation (RMSF) values for the protein and ligand were analyzed to assess their stability and dynamics during the simulation. Additionally, the interactions between the protein and ligand, as well as the ligand’s properties and torsion profile, were also evaluated. The binding energies of the complexes were calculated using GROMACS, a widely-used simulation package, with the CHARMM36 force field and CGenFF force field generator assigned charges to the ligand molecules. The system was neutralized by adding ions to the dodecahedron solvation box with a three-site water solvent model. The potential energy of the docked complex was calculated using molecular mechanics Poisson-Boltzmann surface area (MMPBSA) after equilibrating the system for 1 ns with constant number of particles, volume, and time (NVT). Overall, these analyses provide valuable insights into the stability and dynamics of the protein-ligand complexes and their potential as therapeutic agents.
Results and Discussion
- Molecular targets from SARS CoV-2
This study focuses on essential viral proteins that play a critical role in the entry and replication of the virus. The targets under investigation include the spike receptor binding domain, 3C-like protease (3CLPro), papain-like protease (PLpro), RNA dependent RNA polymerase (RdRp), and membrane protein, each of which is distinguished by its unique properties, as presented in the table below.
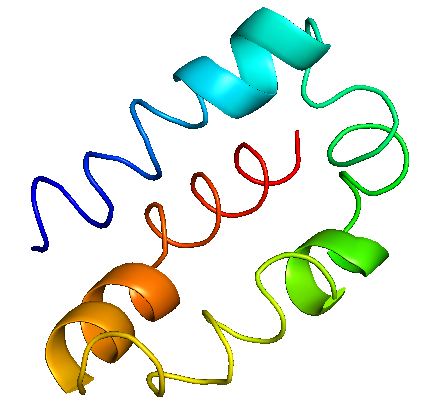
While the three-dimensional structures of the former four proteins were available in their native form on Protein Data Bank (PDB) under the PDB IDs 6M0J, 6LU7, 6W9C, and 6M71, respectively, the membrane protein’s structure was computationally modeled due to the unavailability of its native structure.
The homotrimeric spike glycoprotein, an important protein on SARS CoV-2, facilitates viral entry by binding to cellular receptors, ultimately leading to the fusion of cell and viral membranes. This cell entry process is primarily driven by the spike glycoprotein’s receptor binding domain, which interacts with the ACE2 cell receptor. Afterward, the S1 subunit of the glycoprotein dissociates, and the S2 subunit transitions into its stable post-fusion state to facilitate membrane fusion (Lan et al., 2020). Additionally, 3CLpro is known to play a critical role in mediating viral replication and transcription in coronaviruses (Jin et al., 2020), while PLpro is involved in cleaving the replicase polyproteins’ amino and carbon-end terminals (Osipiuk et al., 2020). The RNA-dependent RNA polymerase, also known as nsp12, is an indispensable component of the coronaviral replication and transcription machinery that catalyzes viral RNA synthesis with the help of cofactors nsp7 and nsp8 (Gao et al., 2020).
With a sequence length of 222 amino acids, the membrane protein is said to be an essential component of the viral envelope that is crucial for virus morphogenesis and assembly. The 3D structure of membrane protein was predicted using trRosetta, which uses deep neural networks in order to build protein structures based on direct energy minimizations (Du et al., 2021). The energy of the model was -11045.1 kJ/mol, which was further minimized to -9027.832 kJ/mol using the YASARA energy minimisation server (Krieger et al., 2009). According to SOPMA, the protein comprises 34.68%, 21.17%, 6.76% and 37.39% of alpha helices, extended strands, beta turns and random coils, respectively. The ERRAT results indicated that the overall quality factor of the model was 96.6019 and the VERIFY3D tool predicted that 57.66% of the residues had a 3D-1D score ≥ 0.2. The Ramachandran plot indicated that 86.1% of the residues were in the most favored region, 11.9% of the residues were in the additional allowed regions, 1.5% of the residues were in the generously allowed regions and 0.5% of the residues were in the disallowed regions. Furthermore, the Z-score predicted by ProSA was found to be -4.34, which indicates the stereo-chemical validation of the protein when compared to that of experimental structures. The membrane protein showed good secondary structure alignment and stereochemical properties, and thus can be considered as a putative drug target. Rephrase this sophistically and creatively
- High throughput screening of natural drug molecules
The structures of the sixty-five drug biomolecules, identified from R. officinalis and A. marmelos, were downloaded from PubChem to check for their drug likeness properties using PreADMET and SwissADME. The results are tabulated below.
The docking results obtained were as follows:

Amoolya Srinivasa
Bioinformatics Programmer at NYU Langone Health